Introduction
Cystic fibrosis is a genetic disorder that occurs due to mutation of the gene called cystic fibrosis transmembrane conductance regulator (CFTR). A mutation in the CFTR affects the transport of sodium and chloride ions across membranes, thus causing production of viscous secretions and thick mucus in organs such as lungs, liver, pancreas, and intestines. The CFTR gene produces a protein that is important in regulating mucus production, sweat, and digestive fluid by regulating the transport of sodium and chloride ions. This means that mutations in the CFTR gene interfere with regulation of water and salt balance in various organs such as lungs, liver, pancreas, and intestines. Although some patients experience intestinal malabsorption, all patients experience lung complications.1 The dominant symptoms of cystic fibrosis manifests in the lungs because the disease causes an increase in mucus secretions, thus causing breathing difficulties. Excessive secretion of mucus encourages infections by pathogens and breathing difficulties, which are the main causes of lung failure and death among patients with cystic fibrosis. Therefore, this research paper seeks to examine etiology, prognosis, radiographic diagnosis, and epidemiology of cystic fibrosis.
Etiology and Prognosis
Cystic fibrosis is a genetic disorder that affects lungs, pancreas, liver, and intestines amongst other organs. Specifically, cystic fibrosis is an autosomal recessive disorder, which occurs due to mutations in the CFTR gene found on chromosome 7 of the human genome.2 A mutation in the CFTR gene or defective CFTR protein causes cystic fibrosis. The CFTR protein is responsible for the transport of chloride and sodium ions across membranes. Hence, mutations in CFTR gene cause defects in expression CFTR protein, which consequently interferes with regulation of salt and water balance in organs such as lungs, liver, pancreas, and intestines. Absence of CTFR protein or its defects causes secretion of thick mucus, which mainly occurs in the lungs when compared with other organs that the disorder affects. Accumulation of thick mucus in the lungs encourages infection of pathogens that ultimately cause permanent lung damage.
Cystic fibrosis is an incurable disorder that mainly affects the lungs. Available treatment options entail alleviation of symptoms of the disorder and improving quality of life of patients. Since cystic fibrosis is a life-threatening disorder with serious social implications, early diagnosis is critical so that patients can receive essential treatment and access critical medical services.3 However, there is marked variation in the way patients respond to various treatment regimes. Usually, the prognosis of cystic fibrosis is dependent on the extent of mutation of the CFTR gene, age of diagnosis, gastrointestinal abnormalities, pulmonary infections, and state of malnutrition.4 Appropriate management of cystic fibrosis increases lifespan of a patient and improves quality of life for treatment reduces progressive damage of organs such as the lungs and intestines. Newborn screening and early treatment have proved to be effective in enabling patients with cystic fibrosis to live quality lives and increase their lifespan into adulthood.
Radiographic Method Used
Radiologists use radiographic methods such as x-ray, computerized tomography, magnetic resonance imaging when diagnosing cystic fibrosis. In this case, the research paper focuses on the use of x-ray in the diagnosis of cystic fibrosis. Since early diagnosis of cystic fibrosis is important in enhancing the management of the genetic disorder, x-ray is one of the radiographic methods that have proved to be indispensable in the diagnosis. Normally, x-ray is applicable in checking the state of the lungs due to cystic fibrosis and pathogenic infections. The x-ray diagnosis examines if pansinusitis is visible in the lungs.5 The appearance of the sinuses indicates the extent of the sinus inflammation. Inflamed sinuses can either mean that an individual is suffering from cystic fibrosis or some pulmonary infections. Hence, genetic test and sweat test can confirm if apparent inflammation of the sinuses is due to cystic fibrosis or other infections.
At the initial stages of cystic fibrosis, it is very difficult to ascertain the nature of infections or condition of the lungs. The x-ray examination of the chest indicates fluffy and diffuse images because fluid and debris accumulate in the alveoli. As cystic fibrosis progress, lungs “whiten out” due to diminished aeration and fibrotic nature of the lungs, which progressively lead to permanent damage to the lungs.6 Hence, keen examination of the x-ray taken can clearly differentiate an inflamed lung due to cystic fibrosis from a normal one. Given that examination of the x-ray is difficult to determine the extent of cystic fibrosis, radiologists usually utilize a standard grading system called Brasfield scoring system.7 The scoring system provides a scale with five parameters each having five points, hence, adding to 25 points altogether. The five parameters are air trapping, linear markings, nodular cystic lesions, large lesions, and generativity. Therefore, assessment of the x-ray film using the parameters provides accurate diagnosis of the severity of cystic fibrosis.
Differential Diagnosis
In x-ray diagnosis, there is a significant difference between a chest of a person with cystic fibrosis and of the one without cystic fibrosis. Although the x- ray films may show similar features, there are subtle features that can differentiate the two films. The nature of sinuses and presence of nasal polyps are two major features that differentiate chest x-ray films of patients with cystic fibrosis and normal people.8 Examination of the sinuses indicates that it appears clear in a normal individual, while in a patient with cystic fibrosis they is extensively grayed area. The extent of the grayed area shows the severity of the panasinusitis, bronchiectasis, and lobar collapse.9 The x-ray picture below indicates that there is an extensive grayed area in the lungs of a patient with cystic fibrosis when compared with that of a normal person. Moreover, the lungs of a patient with cystic fibrosis increase in size due to inflammation of the alveoli and bronchioles. The increase in size is due to accumulation of the mucus and bronchial obstruction of the airways.10 This explains why chest x-ray films of patients with cystic fibrosis appear “whiten out” while those of normal people show clear ribs.
Figure 1 and 2 are x-ray pictures that show chest x-ray of a person without cystic fibrosis and with cystic fibrosis respectively.11
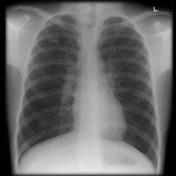
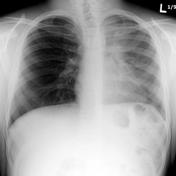
Epidemiology
Cystic fibrosis mainly affects Caucasian population due to their genetic predisposition. Since cystic fibrosis is a genetic disorder, ample evidence indicates that it is common among the whites than the blacks. Statistics show that Hispanics have high incidences of cystic fibrosis of 1 in 9,500 people, while the disorder is very rare among native Asians and native Africans.12 This means that there is racial variation in the occurrence of cystic fibrosis across the world. Additionally, there is gender variation in the diagnosis, prognosis, and treatment of cystic fibrosis. A study in the United States has shown that there is a gender gap in the diagnosis and treatment of cystic fibrosis, which has significant impact on the prognosis of the disorder among women.13 The delayed diagnosis of women in the United States could be due to differential diagnosis of signs and symptoms that male and female patients manifest.
Conclusion
Cystic fibrosis is a genetic disorder that runs through families. The disorder occurs due to the mutations in the CFTR gene, which is found on the chromosome 7 of the human genome. The CFTR gene is responsible for the expression of CFTR protein that aids in the regulation of salts and water balance across membranes. A mutation in the CFTR gene results in expression of defective CFTR protein that is unable to regulate salt and water balance. Although it has no cure, early diagnosis can help in the management of the disorder. Hence, diagnostic methods such as sweat test, genetic test, and radiography are some of the effective methods of diagnosing it. In this case, an x-ray method detects panasinusitis and bronchiectasis. Epidemiological data indicate that cystic fibrosis is prevalent among the whites and mainly affects women more than men due to delay in diagnosis.
Works Cited
DiMuzio, Bruno and Frank Gaillard. “Pulmonary manifestations of cystic fibrosis.” Radiopaedia, 2012. Web.
Lai, Hui-Chuan, Michael Kosorok, Anita Laxova, Linda Makholm, and Philip Farrell. “Delayed Diagnosis of US Females with Cystic Fibrosis.” American Journal of Epidemiology 156.2 (2002): 165-173. Print.
Orenstein, David, Jonathan Spahr, and Daniel Weiner. Cystic Fibrosis: A Guide for Patient and Family. London: Lippincott Williams & Wilkins, 2012. Print.
Wang, Lan, and Steven Freedman. “Laboratory Tests for the Diagnosis of Cystic Fibrosis.” American Journal of Clinical Pathology 117.1 (2002): 109-115. Print.
Whitaker, Kent. Comprehensive Perinatal & Pediatric Respiratory Care. New York: Cengage Learning, 2001. Print.
Endnote
- 1 – Hui-Chuan Lai, Michael Kosorok, Anita Laxova, Linda Makholm, and Philip Farrell. “Delayed Diagnosis of US Females with Cystic Fibrosis.” American Journal of Epidemiology 156.2 (2002): 165.
- 2 – Josee Chariot, and Larry Lands. “Diagnostic Aspects of Cystic Fibrosis.” The Canadian Journal of Diagnosis 2.3 (2001): 94.
- 3 – Lan Wang, and Steven Freedman. “Laboratory Tests for the Diagnosis of Cystic Fibrosis.” American Journal of Clinical Pathology 117.1 (2002): 110.
- 4 – Hui-Chuan Lai, Michael Kosorok, Anita Laxova, Linda Makholm, and Philip Farrell. (2002): 165
- 5 – Lan Wang, and Steven Freedman. (2002):113.
- 6 – Kent Whitaker. Comprehensive Perinatal & Pediatric Respiratory Care. (New York: Cengage Learning, 2001) 437.
- 7 – Bruno DiMuzio, and Frank Gaillard. “Pulmonary manifestations of cystic fibrosis.” Radiopaedia, 2012. Web.
- 8 – David Orenstein, Jonathan Spahr, and Daniel Weiner. Cystic Fibrosis: A Guide for Patient and Family. (London: Lippincott Williams & Wilkins, 2012) 23.
- 9 – Bruno DiMuzio, and Frank Gaillard (2012)
- 10 – David Orenstein, Jonathan Spahr, and Daniel Weiner. (2012) 27.
- 11 – Bruno DiMuzio, and Frank Gaillard (2012)
- 12 – Lan Wang, and Steven Freedman. (2002): 109.
- 13 – Hui-Chuan Lai, Michael Kosorok, Anita Laxova, Linda Makholm, and Philip Farrell. (2002): 165